BASES DE DATOS DE MATERIALES PARA BATERÍAS
El descubrimiento de materiales es clave para la innovación y la comercialización de nuevos productos sostenibles, pero los plazos son del orden de décadas. En los últimos años, el modelizado computacional de materiales ha logrado comprender las relaciones fundamentales entre estructura y propiedades, lo que permite optimizar las propiedades deseadas y diseñar materiales eficaces. Sin embargo, estos estudios suelen emplear cálculos de química cuántica que consumen grandes cantidades de recursos informáticos y son extremadamente costosos en energía. También existe el riesgo de que grupos de investigación independientes que investigan materiales similares repitan estos estudios computacionales, lo que reduce su eficacia.
Un enfoque alternativo que ha ido ganando la atención de la comunidad investigadora se centra en el análisis topológico de las redes de materiales que ya son de dominio público. Estas bases de datos contienen una amplia gama de datos experimentales y/o computacionales. Existen varias bases de datos computacionales de alta calidad sobre materiales y sistemas informáticos asociados, como el Materials Project, AFLOWLIB, NoMaD y la Open Quantum Materials Database (OQMD). Éstas complementan los conjuntos de datos experimentales existentes, como la Base de Datos de Estructuras de Cristales Inorgánicos (ICSD), el Repositorio de Datos de Materiales del NIST o el Pauling File.
Estas grandes bases de datos pueden utilizarse para buscar posibles materiales candidatos, aún no descubiertos, o extrapolar entre materiales para identificar nuevos candidatos y explorar posteriormente su capacidad de síntesis. Este flujo de trabajo podría guiar a los experimentadores en su fabricación eficiente. Se trataría de una alternativa al enfoque de ensayo y error, que suele ser muy exigente en términos de tiempos y costes de síntesis.
Por supuesto, siguen existiendo ciertas advertencias, en particular la de garantizar que los datos recogidos sean lo suficientemente precisos; de lo contrario, los patrones identificados en los datos corren el riesgo de ser ficticios. De hecho, esto se aplica a todos los enfoques basados en datos, por lo que hay que tener cuidado de comparar y verificar los conjuntos de datos con el experimento.
REDES DE MATERIALES PARA BATERÍAS
El análisis topológico de las redes de materiales para identificar materiales que aún no se han descubierto tiene dos retos teóricos. El primero es identificar compuestos termodinámicamente estables, lo que también se denomina problema de predicción de estructuras. Y el segundo es su sintetizabilidad, que suele implicar la evaluación de estructuras metaestables, tiempos de vida y energías de reacción. En los últimos 20 años, el desarrollo de la química computacional ha permitido predecir no sólo el estado básico más estable, sino también las estructuras metaestables de baja energía. Esto ha llevado a la identificación de un número creciente de nuevos materiales hipotéticos. En esencia, las consideraciones termodinámicas reducen el espacio químico en el que los experimentadores deben buscar, y este conocimiento puede utilizarse para predecir la probabilidad de síntesis de estas hipotéticas estructuras estables y metaestables.
El reto de la sintetizabilidad es formidable. Hay que encontrar una ruta que conduzca al compuesto deseado a partir de una multitud de reactivos y productos candidatos, todo ello teniendo en cuenta las reacciones de varios pasos, las reacciones secundarias, la posibilidad de compuestos metaestables y las barreras de los estados de transición. Sin embargo, se están haciendo avances, y el grupo de Modelizado y Simulación Computacional del CIC energiGUNE pretende emplear la ciencia de redes en un enfoque basado en gráficos para abordar este problema. El enfoque utiliza los nodos y enlaces de las redes para trazar las relaciones termodinámicas entre los materiales y luego identificar posibles rutas sintéticas. Este enfoque permitiría guiar a los experimentadores y maximizar sus recursos para acelerar la fabricación de nuevos materiales y su desarrollo.
Uno de estos recursos es la base de datos OQMD de materiales, recientemente desarrollada, que codifica los datos de estabilidad termodinámica de muchos compuestos inorgánicos. A medida que se añaden más datos a esta red, tiene la posibilidad de evolucionar y verificarse, ya que los agujeros en la red pueden identificar materiales aún por descubrir. Estas bases de datos distan mucho de ser completas; de hecho, debido al gran número de combinaciones posibles de elementos que pueden combinarse para formar un compuesto, es imposible obtener una base de datos completa. Sin embargo, siguen evolucionando y tienen la oportunidad de validarse al predecir con éxito materiales aún no descubiertos. La analogía es similar a las lagunas o agujeros en la tabla periódica que predijo Mendeléyev en 1869 y que, posteriormente, se validaron con el descubrimiento del galio en 1875.
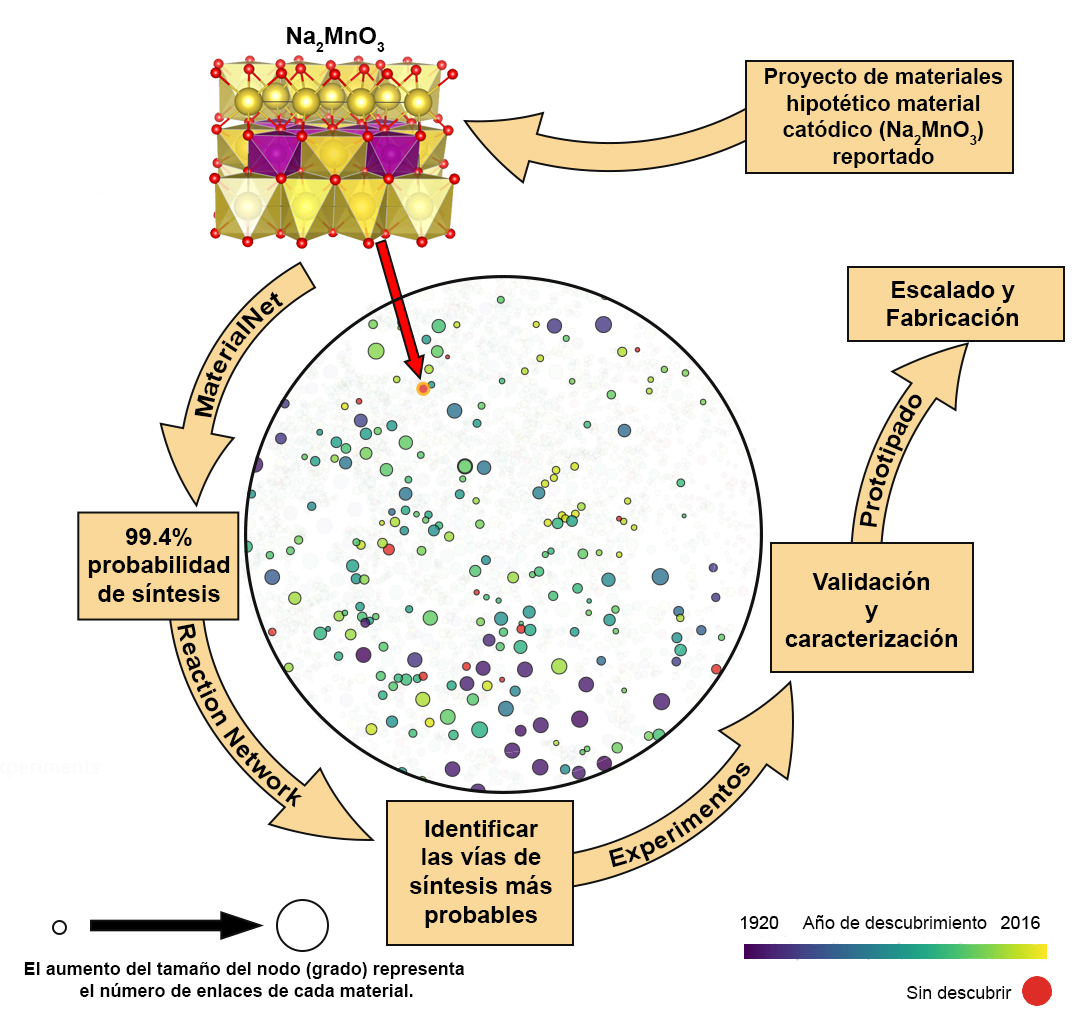
Ahora incluso es posible visualizar las redes de materiales utilizando la base de datos en línea Materials Similarity Network. Por ejemplo, tomemos la red local del hipotético material no descubierto Na2MnO3 (véase Aziz y Carrasco), del que se han predicho propiedades prometedoras y del que se espera una probabilidad de síntesis del 99,4%. El siguiente paso en este enfoque descendente sería la identificación de posibles rutas de síntesis a través de una Red de Reacción, seguida de una validación experimental. La identificación de posibles rutas de síntesis ayudaría a los experimentadores a reducir el número de rutas de reacción a considerar, incluso si la red finalmente no lograra predecir la ruta de reacción óptima.