MATERIAL DATABASES FOR BATTERIES
Material discovery is key to the innovation and commercialization of new sustainable products, but the timescales involved are on the order of decades. Over the last years, computational modeling of materials has succeeded in understanding fundamental structure-property relationships, enabling the optimization of desired properties and effective material design. However, these studies often employ quantum chemistry calculations that consume large amounts of computer resources and are extremely energy-intensive. There is also the risk that independent research groups investigating similar materials repeat these computational studies reducing efficiencies.
An alternate approach that has recently been gaining the research community´s attention focuses on the topological analysis of material networks that are already in the public domain. These databases contain a vast array of experimental and/or computational data. There are several high-quality computational materials databases and related informatics, such as the Materials Project, AFLOWLIB, NoMaD, and the Open Quantum Materials Database (OQMD). These complement existing experimental data sets such as the Inorganic Crystal Structure Database (ICSD), NIST Materials Data Repository, or the Pauling File.
These large databases can be used to search for potential candidate materials, not yet discovered, or extrapolate between materials to identify novel candidates and subsequently explore their synthesizability. This workflow could guide experimentalists in their efficient fabrication. This would be an alternative to a trial-and-error approach that usually has high demands in terms of synthesis times and costs.
Of course, certain caveats remain, particularly ensuring collected data is of sufficient accuracy; otherwise, identified patterns in the data risk being fictitious. In fact, this applies to all data-driven approaches, so care must be taken to benchmark and verify datasets with the experiment.
MATERIAL NETWORKS FOR BATTERIES
The analysis of materials networks to identify materials that have yet to be discovered has two theoretical challenges. The first is to identify thermodynamically stable compounds, also referred to as a structure prediction problem. And the second is their synthesizability, which typically involves evaluating metastable structures, lifetimes and reaction energies. Over the last 20 years, the development of computational chemistry has enabled the prediction of not only the most stable ground state but also low-energy metastable structures. This has led to the identification of an increasing number of new hypothetical materials. In essence, thermodynamic considerations narrow down the chemical space in which experimentalists should look, and this knowledge can be used to predict the synthesis probability of these hypothetical stable and metastable structures.
The synthesizability challenge is formidable. One needs to find a route that leads to the desired compound from a multitude of candidate reactants and products, all while considering multi-step reactions, side reactions, the possibility of metastable compounds and transition state barriers. However, inroads are being made, and the Modeling and Computational Simulation group at CIC energiGUNE aims to employ network science in a graph-based approach to address this issue. The approach uses the nodes and links of networks to map the thermodynamic relationships between materials and then identify possible synthetic routes. This approach would guide experimentalists and maximize their resources to accelerate the fabrication of new materials and development.
One such resource is the recently developed materials OQMD database encoding thermodynamic stability data for many inorganic compounds. As more data is added to this network, it has the scope to evolve and verify itself as holes in the network may identify materials yet to be discovered. These databases are far from complete; in fact, due to the sheer number of possible combinations of elements that can be combined to form a compound, it is impossible to obtain a complete database. However, they continue to evolve, and they have the opportunity to validate themselves by successfully predicting undiscovered materials. The analogy is similar to the gaps or holes in the periodic table predicted by Mendeleev in 1869, and subsequently, validated by the discovery of gallium in 1875.
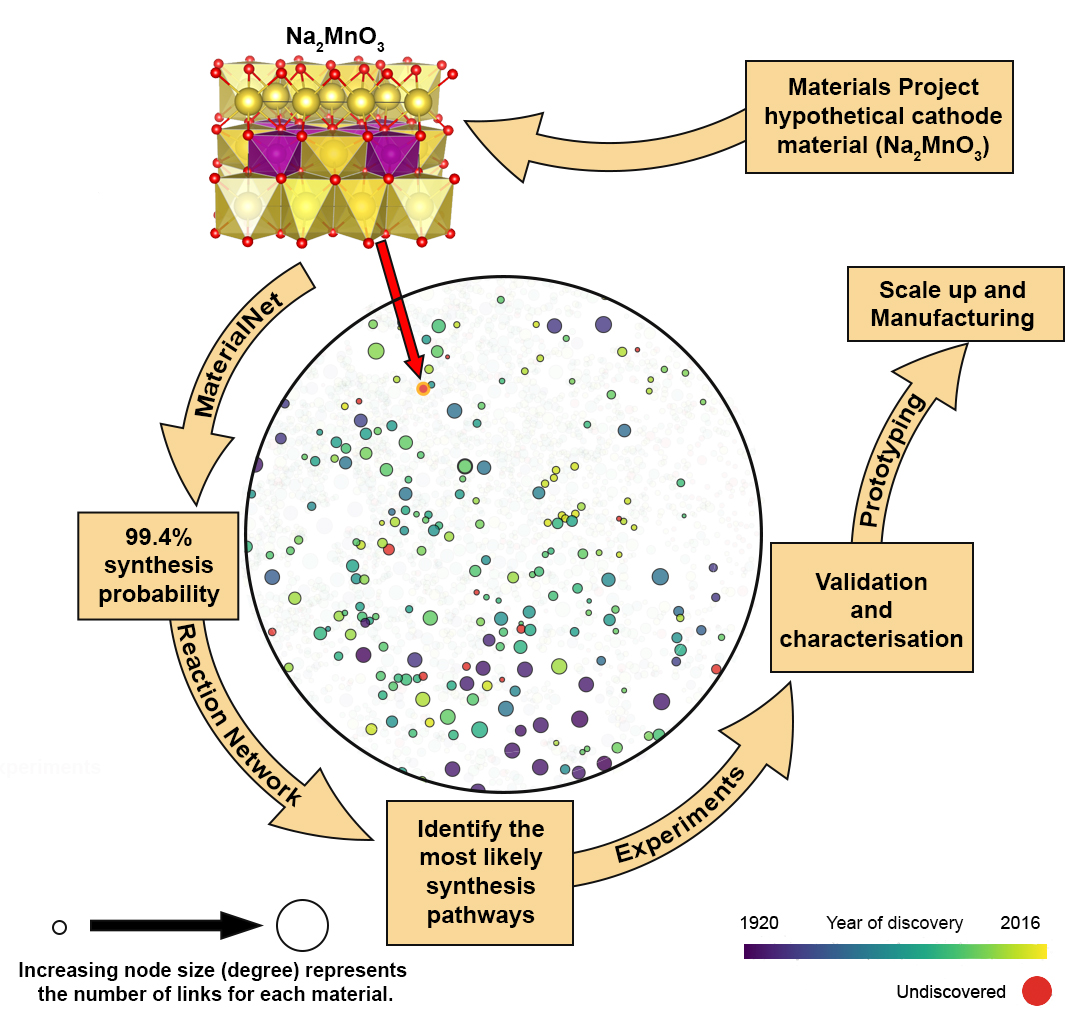
It is now even possible to visualize material networks using the online Materials Similarity Network database. For example, take the local network for the hypothetical undiscovered material Na2MnO3 (see Aziz & Carrasco) that has been predicted to have promising properties and is expected to have a synthesis probability of 99.4%. The next step in this top-down approach would be identifying possible synthesis pathways through a Reaction Network, followed by experimental validation. Identifying possible synthesis routes would help experimentalists reduce the number of reaction pathways to consider, even if the network ultimately failed to predict the optimal reaction pathway.