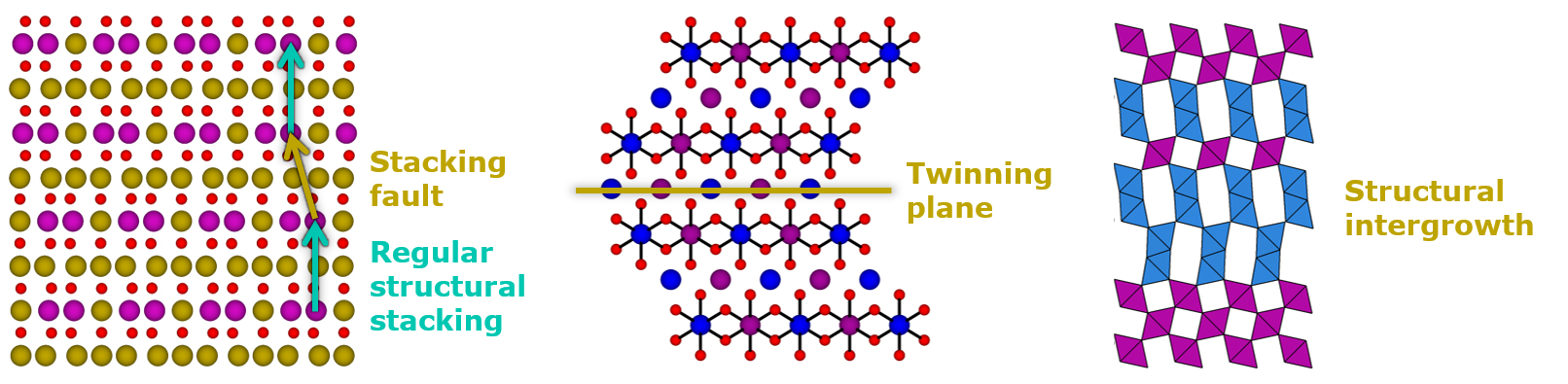
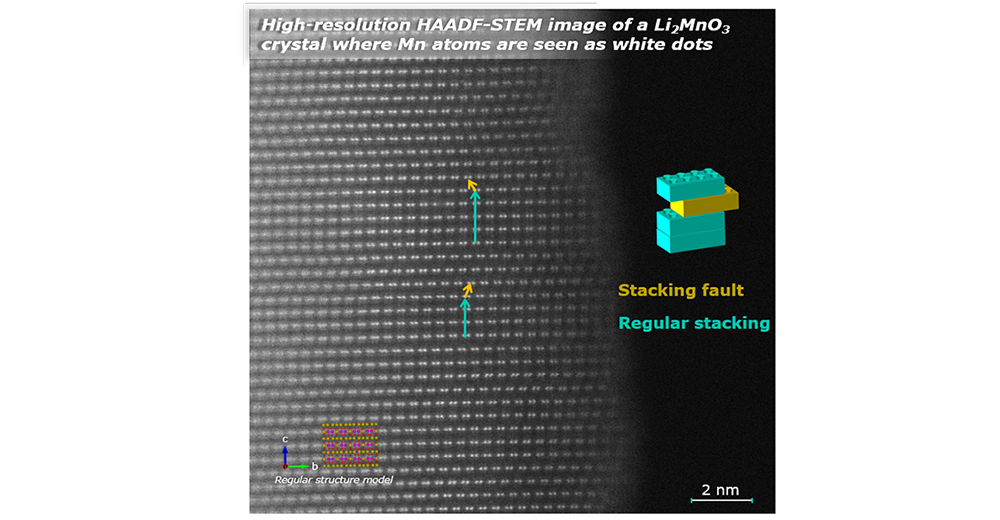
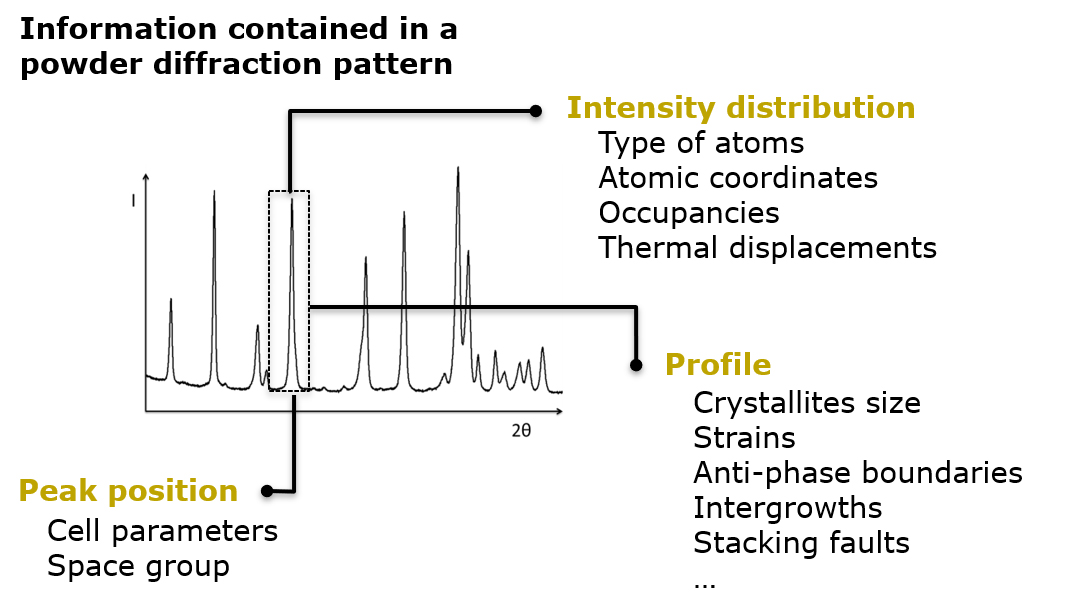
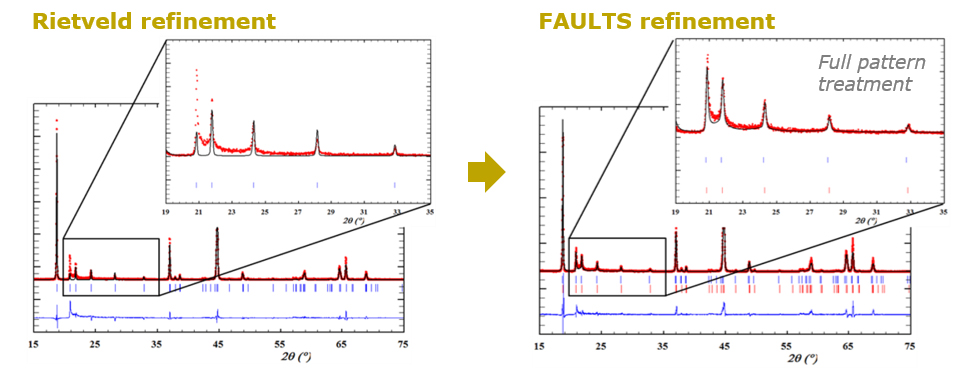
La imperfección es una característica universal en los materiales cristalinos
"Los cristales son como las personas: ¡son los defectos en ellos los que tienden a hacerlos interesantes!" Esta cita del Prof. Collin Humphreys, físico especialista en semiconductores (Universidad de Cambridge), también es válida para los materiales de las baterías. Sin embargo, la presencia de defectos en sus estructuras cristalinas es a menudo ignorada o simplificada en exceso, por falta de medios para caracterizarlas adecuadamente.
Los cristales se definen generalmente como sólidos en los que los elementos constitutivos se ordenan en un patrón específico que se repite periódicamente en las tres dimensiones del espacio. Esa definición puede sugerir que los materiales cristalinos (incluida la mayoría de los materiales electroactivos para las baterías de iones de litio y de sodio-ion) son distribuciones perfectas de los átomos, pero en realidad, la imperfección es una característica universal de los materiales cristalinos.
Los defectos cristalinos generalmente tienen mala reputación. Por sí mismos, los nombres "defecto" y "fallo" tienen una connotación claramente negativa. En consecuencia, se consideran, a priori, características perjudiciales para las propiedades y el rendimiento de los materiales funcionales y, a su vez, se pueden hacer esfuerzos considerables para reducir al mínimo su concentración o contrarrestar su impacto perjudicial.
Sin embargo, los defectos pueden generar valor. Los defectos e impurezas que causan el color altamente deseable en los diamantes de calidad gema son de hecho una ilustración manifiesta de la oportunidad que ofrecen las imperfecciones estructurales. Cuando se les cambia el nombre a "dopantes", los defectos empiezan a ser más atractivos. Toda la industria de los semiconductores se basa en métodos para preparar materiales de Si, Ge o GaAs dopados con cantidades precisas de las impurezas deseadas, lo que permite afinar sus propiedades eléctricas.
Por lo tanto, en la Ciencia de los Materiales, en lugar de evitar los defectos, la introducción intencionada y deliberada de defectos (con control sobre el tipo, la concentración y la ubicación) ofrece nuevas oportunidades para la ingeniería de materiales. El científico especializado en materiales puede entonces utilizarlas como una herramienta para afinar y mejorar las propiedades e incluso permitir nuevas funcionalidades.
En el campo de los materiales para baterías, el desorden y los defectos estructurales también son omnipresentes. El reto consiste en caracterizar con precisión la microestructura de los compuestos electroactivos, comprender las correlaciones entre las características microestructurales de las muestras y su rendimiento, y eventualmente controlar la ubicación y cantidad de los defectos durante la síntesis y/o procesamiento de los materiales, para convertirlos en una ventaja y mejorar sus propiedades. Este es el trabajo del Químico del estado sóilido y CIC energiGUNE cuenta con algunos expertos reconocidos internacionalmente en este campo.